- 20 min read
- Life sciences
- Sep 2025
Building a prototype for One-Click drug discovery

One-Click drug discovery
We built a prototype end-to-end ML system that demonstrates how drug discovery workflows could be streamlined into a computational workflow. Starting from a disease name, it identifies relevant biological targets, learns from real bioactivity data, and ranks candidate molecules by predicted activity. While not a tool for finding real drugs without substantial experimental validation, it illustrates how AI can structure and accelerate the discovery process.
Background
Drug development is one of the most expensive, time-intensive, and uncertain processes in modern science.
- Timeline: Bringing a new medicine from discovery to market typically takes 10-15 years [1].
- Cost: The average R&D cost per new drug falls between $314 million and $2.8 billion, depending on methodology and assumptions [2].
- Success rate: Only about 10-20% of drug candidates that enter clinical trials ultimately secure approval [3].
These long timelines and high failure rates lead to delayed treatments, enormous healthcare costs, and increased suffering worldwide.
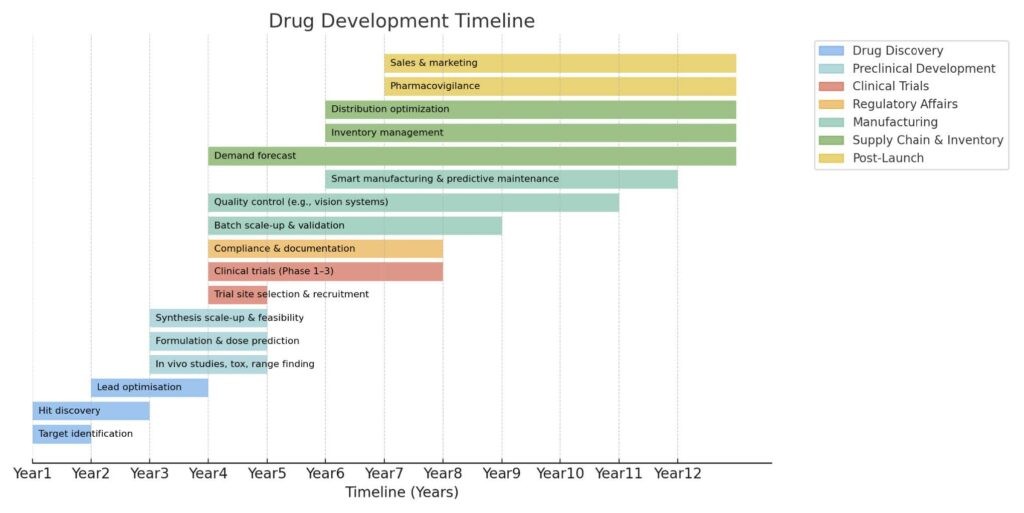
The traditional drug development pipeline (Figure 1) begins with discovery and preclinical work, progresses into clinical trials, and integrates regulatory, manufacturing and supply chain planning before extending into post-launch pharmacovigilance and commercialization.
While every stage is important, the earliest stage – drug discovery – plays a vital role in determining overall success. When discovery fails, the effects multiply across every later stage, making it the biggest bottleneck in the pipeline.
Drug discovery
The discovery phase is where every drug development program begins. Its goal is to identify biological targets linked to a disease, and chemical compounds that can act on them. In practice, this stage involves:
- Target identification: Determining which proteins, genes, or pathways are involved in the disease.
- Hit discovery: Screening millions of compounds to find a small fraction that show measurable activity (“hit”).
- Lead optimization: Iteratively refine chemical structures to improve potency, safety and drug-like properties.
Key challenges in drug discovery include:
- High failure in target identification: A false discovery rate of around 92% has been reported for preclinical target selection [4], meaning that most proposed disease-protein links do not hold up under validation.
- Extremely low hit rates: Large-scale compound screens often yield hit rates of less than 1% [5].
- Intuition-driven chemistry: Lead optimisation often relies heavily on expert judgement and trial-and-error, making progress slow and difficult to scale.
- Sequential workflows: Each cycle of design, synthesis, and testing is performed in series rather than in parallel, stretching timelines across years.
These challenges make drug discovery particularly well suited to improvements through modern data-driven approaches. Advances in machine learning (ML) and computational biology now provide tools that can support and accelerate early-stage research.
To illustrate how this might look in practice, we implemented a prototype one-click pipeline that finds promising compounds to known diseases.
Technical exploration: A Prototype discovery pipeline
System Overview
At a high level, the pipeline integrates structured biomedical knowledge, curated bioactivity data, and ML models into a single automated workflow.
Starting with a disease name, the system identifies the most promising biological targets, trains predictive models of compound activity, expands candidate space via large-scale screening, and ranks compounds by predicted activity (Figure 2).
At a high level, the pipeline integrates structured biomedical knowledge, curated bioactivity data, and ML models into a single automated workflow.
Starting with a disease name, the system identifies the most promising biological targets, trains predictive models of compound activity, expands candidate space via large-scale screening, and ranks compounds by predicted activity (Figure 2).

Prototype run setup
To make the pipeline tangible, we executed a proof-of-concept run using our development codebase, built collaboratively by our ML engineer and Claude code [6], an AI coding assistant.
For this demonstration, we restrict the virtual screening to 1 million compounds from ZINC20, rather than the full database. The run completed in ~33 minutes on a single CPU core, whereas processing the entire ZINC20 drug-like collection (≈288 million compounds) would require several days on a single core, or roughly two days even with 8-core parallelization.
The run was launched with the following command:
python -m drug_discovery.cli disease=”Type 2 Diabetes”
pipeline.target_identification.max_targets=50
pipeline.lead_expansion.max_zinc_compounds=1000000Which initialized the pipeline as:
🧬 Drug Discovery Pipeline
==================================================
Disease: Type 2 Diabetes
Targets searched: 50
Max compounds: 1,000,000The logs in the following sections illustrate how the system steps through disease target selection, data curation, model training, and virtual screening.
Stage 1: Automated target identification
Every drug discovery project starts with a simple but critical question: what part of human biology should we target? This usually means selecting a biological target – most often a protein such as an enzyme or receptor – that plays a role in disease.
Traditionally, researchers choose targets by manually reviewing literature, piecing together experimental results, and drawing on expert judgment [7]. While this has produced many important discoveries, it is inherently limited by human bias and the difficulty of integrating the vast and constantly expanding biomedical knowledge base.
In our prototype, we leveraged the Open Targets platform [8], a widely-used resource that aggregates evidence from genetics, clinical trial data, biomedical literature, and pathway databases to link diseases with potential therapeutic targets.
Given only a disease name, the system retrieves all associated targets.
Note
📦 How specific should the disease name be?
The pipeline accepts anything from broad terms (“cancer”) to specific diseases (“breast cancer”) or even subtypes when available. In practice, more specific inputs yield more actionable targets.
- With “cancer”, the top hits include universal cancer genes (BRCA1/2, TP53) that are central to biology but rarely directly drugged with small molecules, alongside general kinases like EGFR or KRAS, which are technically druggable but not always the most relevant target for a specific cancer type.
- With “breast cancer”, the list shifts toward breast-relevant signaling proteins such as PIK3CA, ESR1, and ERBB2, which have clearer roles in this disease and a long track record of tractable drug mechanisms.
The system then filters these targets by two criteria:
- Association strength – how strong the evidence is for the target’s role in the disease.
- Data availability – whether enough bioactivity measurements exist in ChEMBL [8] – a large-scale bioactivity database that will serve as our QSAR modelling dataset in later stages.
Note
📦 Known vs. novel targets?
In this prototype, the pipeline picks well-studied targets – the ones with lots of existing bioactivity data. These are often proteins that already have drugs on the market or in development. However, it doesn’t mean they are used up: new generations of drugs are often made for the same target to improve safety, reduce side effects, or overcome resistance.
On the other hand, novel targets come from exploratory science, for example:
- Genetics – linking specific gene mutations in patients to disease risk.
- Omics profiling – scanning across all DNA, RNA, or proteins to spot unexpected disease-related patterns.
- Phenotypic screens – testing compounds on cells or tissues, then later deconvolving which protein the active ones hit.
In practice, both known and novel targets matter: known ones let you build better drugs faster, while novel ones open doors to entirely new therapies.
This dual filtering ensures we select targets that are both biologically promising and have sufficient experimental data for model training.
[Stage 1: Automated Target Identification]
→ Querying Open Targets API for disease Type 2 Diabetes (MONDO_0005148)
→ Found 25 associated targets (cap = 50)
→ Cross-checking with ChEMBL for IC₅₀ assay coverage:
– KCNJ11 (assoc. score 0.867): 0 IC₅₀ activities → ❌ skipped (no usable data)
– ABCC8 (assoc. score 0.864): 3 IC₅₀ activities → ❌ skipped (too sparse)
– GCK (assoc. score 0.862): 85 IC₅₀ activities → ❌ skipped (too sparse)
– PPARG (assoc. score 0.849): 1,607 IC₅₀ activities → ✅ selected
→ Selected PPARG (CHEMBL235) with rich activity datasetStage 2: Data Curation
Once a target is chosen, the next step is to gather experimental data that describes how different molecules interact with it. This bioactivity data provides the ground truth for training predictive models.
Most of these measurements are reported as IC₅₀ values, a standard way of describing how strongly a compound inhibits a target.
Raw data from sources like ChEMBL [9] is invaluable, but it is often noisy, reported in inconsistent units, contains duplicate or contradictory measurements, and some values may be extreme outliers. Without careful curation, these issues can reduce model reliability.
Our pipeline automates the entire curation process:
- Query ChEMBL data: Retrieves all bioactivity records for the selected target.
- Standardize units: Convert IC₅₀ values to a common nanomolar scale.
- Transform potency: Convert to pIC₅₀ values, a negative log-scale potency, where higher values indicate a compound is more potent.
- Aggregate duplicates: When a compound has multiple entries, take the median for stability.
- Filter noise: Keep only values within a biologically reasonable range to reduce the impact of experimental noise and outliers.
Note
📦 What is IC₅₀ really?
Think of IC₅₀ as a “dose strength test.” It represents the concentration at which a compound reduces the target’s activity by half.
- Low IC₅₀ – potent (works at very small amounts)
- High IC₅₀ – weak (needs much more to have an effect)
pIC₅₀ is just the negative log of IC₅₀. Because it’s on a log scale:
- Higher pIC₅₀ – more potent compound
- Lower pIC₅₀ – less potent compound
[Stage 2: Data Curation]
→ Retrieved 1,457 unique compounds from ChEMBL
→ Final dataset: 1,456 molecules with valid pIC₅₀ valuesStage 3: QSAR modelling
Each biological target identified in Stage 1 receives its own predictive model, trained on curated data relevant to the target.
In drug discovery, this task is known as Quantitative Structure–Activity Relationship (QSAR) modeling. The model’s purpose is simple: given a new compound, predict how strongly it will interact with the chosen biological target, allowing us to virtually “test” millions of compounds without running a single experiment in the lab.
To represent molecules numerically, we start from SMILES strings – a compact text notation for chemical structures. An example of a SMILES representation is shown in Figure 3b.
Note
📦 What’s a SMILES string?
SMILES (Simplified Molecular Input Line Entry System) is a way to write down molecules using text instead of drawings. For example, water is written as O, and ethanol as CCO. It’s like shorthand chemistry that computers can easily process.
Using RDKit [10], an open-source cheminformatics toolkit, we convert these SMILES into a set of 64 molecular descriptors, which capture:
- Size and composition, e.g., molecular weight and how many atoms of different types it contains.
- Physicochemical properties, such as lipophilicity (how oily a molecule is) and polarity (how it interacts with water).
- Structural features, like the number of rings, whether they are aromatic, and how flexible the molecule is.
- Drug-likeness heuristics, which predict whether a molecule is likely to be orally active in humans.
Together, they provide a structured fingerprint of each molecule that the model can learn from.
For prediction, we train an XGBoost regressor [11], with hyperparameters optimized via Optuna [12] and model robustness evaluated through 3-fold cross-validation.
This target-specific predictive model provides the foundation for the next stage, where we expand into new chemical space and search for promising candidate compounds beyond the training data.
[Stage 3: QSAR Modelling]
→ Generated 64 RDKit descriptors
→ Training model with Optuna (200 trials)
→ Best CV R²: 0.560 ± 0.018 | Training R²: 0.893
→ Top influential features: [‘aromatic_rings’, ‘logp_lipinski’, ‘num_aliphatic_heterocycles’, ‘aliphatic_rings’, ‘mw_lipinski’, ‘chi0v’, ‘num_saturated_carbocycles’, ‘num_saturated_heterocycles’, ‘bertz_ct’, ‘slogp_vsa1’]Stage 4: Similarity-based lead expansion
The next step is to search for new candidate molecules beyond the compounds already measured experimentally in ChEMBL. In traditional drug discovery, this is often done using high-throughput screening (HTS), physically testing very large numbers of compounds in the lab to see which show activity against the target.
Note
📦 Why not just brute-force test everything?
HTS can screen millions of compounds, but it is expensive, time-consuming, and typically yields low hit rates. Virtual expansion flips the approach: instead of testing blindly, start with a few compounds already known to work, then search for close chemical cousins that are more likely to succeed.
In our pipeline, we conduct virtual lead expansion. Instead of testing millions of molecules blindly, we start from a set of known active compounds (the “leads”), and use them as templates to search a large library ZINC20 [13], which provides millions of commercially available molecules, for structurally similar molecules. This approach is grounded in the well-established similar property principle: molecules with similar chemical structures are likely to behave alike in biological systems [14,15].
To make this robust, the expansion process includes several safeguards and filters:
- Selecting lead templates: From the ChEMBL data, we automatically extract the most potent compounds against the target – for example, those with pIC₅₀ ≥ 7, compounds potent enough to inhibit the target at 100 nanomolar or lower concentrations.
- Excluding training compounds: Before screening ZINC20 compounds, we exclude any molecule already present in the CHEMBL training set. This prevents data leakage and ensures we are not “rediscoverying” what the model has already seen.
- Drug-likeness prefiltering: From ZINC20, we keep only molecules that satisfy common medicinal chemistry rules, Lipinski’s Rule of Five [16]. Molecules that are too large, too greasy, or too polar often struggle to be absorbed or distributed properly in the body. Therefore, these rules check that molecules have reasonable molecular weight, lipophilicity, and limited numbers of hydrogen bond donors and acceptors. Such filters are widely used because compounds that break too many of these rules tend to fail as drugs in later stages of development.
- Similarity calculation: To compare molecules, we use Morgan fingerprints [17] to generate structured digital representations of their features. An example of Morgan fingerprint is illustrated in Figure 3c. These encodings are then compared using the Tanimoto similarity coefficient [18], which quantifies how much two molecular fingerprints overlap. A threshold (e.g. ≥0.4) balances inclusivity and selectivity, keeping molecules related to the known leads while still leaving room to explore new chemical space.
Note
📦 What is Lipinski’s Rule of Five?
A simple rule of thumb used in medicinal chemistry to flag whether a compound is likely to be orally active in humans. It’s called the “Rule of Five” because the cutoffs are all multiples of five:
- ≤ 5 hydrogen bond donors
- ≤ 10 hydrogen bond acceptors (≈ 2×5)
- Molecular weight < 500 Daltons
- logP (lipophilicity) ≤ 5
Compounds that obey these rules are more likely to be absorbed when taken by mouth. They’re not strict laws – many drugs break them – but they’re a useful first filter when working with large libraries.
📦 How do fingerprints and similarity work?
- Morgan fingerprints: These are circular substructure encodings. They look at each atom and its neighborhood (local chemical environment) and encode it into a digital map. That makes them especially powerful for detecting structural similarities.
- Tanimoto score: Imagine comparing two such maps. A score of 1.0 means identical, 0.0 means no overlap. Scores in between capture varying degrees of resemblance.
The result of this stage is a filtered set of ZINC20 molecules, that are novel (not in training), drug-like, and structurally related to known active leads. These candidates are then passed forward for prioritization in Stage 5.
[Stage 4: Similarity-Based Lead Expansion]
→ Loaded 1,000,000 compounds from ZINC20
→ Excluded 1,457 training set molecules to prevent data leakage
→ Extracted 582 potent lead templates (pIC₅₀ ≥ 7.0, range: 8.70–10.22) from ChEMBL
→ Filtered to 990,444 drug-like molecules with valid SMILES
→ Generated RDKit fingerprints for 50 leads; applied Tanimoto similarity threshold ≥ 0.4
→ 90 structurally similar candidates found (similarity: 0.400 – 0.618; mean ~0.44)Stage 5: Scoring and prioritization
The next step is prioritization: with a filtered pool of candidate molecules, which ones should move forward to experimental validation?
In this prototype, we rank compounds by predicted activity. The QSAR model from Stage 3 estimates the compound’s potency against the target: higher predicted pIC₅₀ values indicate stronger inhibition or binding.
However, potency alone isn’t enough. A compound that looks strong on paper is of little use if chemists cannot actually make it in the lab. This is why synthetic accessibility is important. To provide this context, we calculate a composite synthesis score, although it is not used directly in the ranking. Using RDKit [10], we combine several signals:
- Synthetic Accessibility Score (SAScore) – a widely used estimate of ease of synthesis.
- Retrosynthetic difficulty – how many steps are likely required to build the compound from purchasable fragments.
- Structural complexity – unusual scaffolds, lots of rings or stereocenters.
- Fragment analysis – favoring molecules made from common building blocks.
By ranking compounds on predicted activity while showing synthetic accessibility as a reference, the results remain transparent and easy to interpret. Future versions could integrate multiple objectives into a weighted scoring scheme, but here we deliberately keep the scoring simple.
For validation, the system compares the top predicted ZINC candidates against experimentally confirmed ChEMBL compounds, highlighting potency gaps and synthesis trade-offs.
[Stage 5: Scoring and Prioritization]
→ ZINC20 lead expansion completed: 50 top candidates identified
→ Calculated synthesis scores for all candidates
→ Ranked 50 molecules by predicted activity (synthesis scores shown as reference)
→ Best predicted pIC50: 7.54
→ Compared top ZINC candidates vs top ChEMBL training compounds:
– Avg Top-10 ChEMBL (experimental): pIC50 = 9.66 | Avg synthesis score = 0.695
– Avg Top-10 ZINC (predicted): pIC50 = 7.02 | Avg synthesis score = 0.758
– Insight: ZINC predictions lower in potency, but potentially easier to synthesize
🏆 Top 3 ZINC Candidates:
————————————————–
1. ZINC560983256 | Pred pIC50: 7.54 | Synth Score: 0.720
2. ZINC65094916 | Pred pIC50: 7.27 | Synth Score: 0.763
3. ZINC65107816 | Pred pIC50: 7.25 | Synth Score: 0.761Analysis of the top candidate (ZINC560983256)
A key question is: Are our new candidates competitive with the best compounds already known? To find out, we compare the highest-ranked ZINC candidate against both top ChEMBL compounds and marketed PPARG drugs (Table 1).
The ChEMBL top-10 compounds achieve exceptional potency (average pIC₅₀ 9.66). However, none of the approved PPARG drugs – pioglitazone, rosiglitazone, lobeglitazone – appear in the top-100 ChEMBL list at all. This highlights an important point: the most potent compounds in databases are not necessarily the ones that become medicines. Many highly potent compounds fail later in development due to safety issues (e.g., side effects), unwanted interactions with other proteins, or poor behavior in the body (such as poor absorption or rapid breakdown) [19]. In contrast, marketed PPARG drugs accept a more modest potency profile but succeed because they balance activity with acceptable safety and reliable performance in patients.
Our top ZINC candidate (ZINC560983256) has the following properties:
| Group | Compounds | Approval Year | pIC50 | Synth Score |
|---|---|---|---|---|
| Marketed Drug | Pioglitazone | 1999 (US, global) | 5.76 | 0.80 |
| Marketed Drug | Rosiglitazone | 1999 (US, global) | 6.80 | 0.77 |
| Marketed Drug | Lobeglitazone | 2013 (South Korea) | 7.74 | 0.69 |
| Top-10 ChEMBL (AVG) | N/A | N/A | 9.66 | 0.70 |
| ZINC Candidate | ZINC560983256 | N/A | 7.54 | 0.72 |
The ChEMBL top-10 compounds achieve exceptional potency (average pIC₅₀ 9.66). However, none of the approved PPARG drugs – pioglitazone, rosiglitazone, lobeglitazone – appear in the top-100 ChEMBL list at all. This highlights an important point: the most potent compounds in databases are not necessarily the ones that become medicines. Many highly potent compounds fail later in development due to safety issues (e.g., side effects), unwanted interactions with other proteins, or poor behavior in the body (such as poor absorption or rapid breakdown) [19]. In contrast, marketed PPARG drugs accept a more modest potency profile but succeed because they balance activity with acceptable safety and reliable performance in patients.
Our top ZINC candidate (ZINC560983256) has the following properties:
SMILES: CCc1nc2cc(NC(=O)c3cn(CCC#N)nc3-c3ccc([N+](=O)[O-])cc3)ccc2o1
Predicted pIC₅₀: 7.54
Synthesis score: 0.720Key structure features
- Amide bond (NC(=O)): acts like a connector between two parts of the molecule.
- Cyanopropyl group (CCC#N): a side chain ending in a nitrile. Nitriles are often used in real drugs because they can form polar interactions with proteins and sometimes improve metabolic stability [20].
- Nitro group ([N+](=O)[O-]): a common “structural alert” in drug design, frequently linked to mutagenicity and toxicity risks [21].
Pros
- Moderate ease of synthesis: the score of 0.72 suggests a chemist could potentially make it in the lab without excessive difficulty.
- Potentially stabilizing side chain: the cyanopropyl group is a known feature in many marketed drugs and may help the molecule interact with its target [20].
- Distinct chemistry: different layout from approved PPARG drugs, which could enable new binding possibilities.
Cons
- Weaker predicted potency comparing to top ChEMBL compounds: the best PPARG compounds in ChEMBL reach pIC₅₀ of 9–10, while this one is 7.5.
- Toxicity concern: nitro group is often avoided because it can cause safety issues [21].
- No track record for PPARG: while novel, this kind of structure hasn’t been validated for this target, making its translational potential uncertain.
Takeaways
ZINC560983256 does not match the potency of the top ChEMBL compounds, but it falls within the same range as marketed PPARG drugs while remaining synthesizable. This makes it a plausible starting point for exploration. Still, concerns such as the nitro group and the absence of experimental validation mean it would likely require significant optimization and testing before prioritization.
This was a proof-of-concept run, limited to 1 million ZINC20 compounds out of the full 288 million. Even at this scale, the pipeline identified candidates comparable to marketed drugs in terms of activity and synthesizability. Scaling to the full chemical space could uncover stronger ones, but expert review remains essential before moving toward experiments.
Future directions
This study is designed as a proof-of-concept – a way to show that drug discovery workflows can be captured in an automated, modular pipeline. While it highlights the potential of AI-assisted discovery, there are many ways the system could be extended and improved:
- Richer molecular representations: Instead of relying only on predefined descriptors, graph neural networks (GNNs) [22] can directly operate on molecular graphs, learning expressive features that capture structural subtleties missed by handcrafted descriptors.
- Exploring novel chemical space: Our pipeline searches for candidates structurally similar to known actives, but generative models (e.g., variational autoencoders or diffusion-based molecule generators) could propose entirely new molecules, going beyond the constraints of existing databases.
- Integrating toxicity and ADMET models: A compound’s activity, drug-likeness and synthesis accessibility are only part of the picture. Absorption, distribution, metabolism, excretion, and toxicity (ADMET) [23] profiles often determine whether a drug fails in the clinic. Adding toxicity and safety predictors would make the scoring more realistic.
- Multi-target optimization: Many diseases involve complex networks of pathways, and effective therapies may need to modulate several targets simultaneously [24]. Extending the pipeline to multi-target QSAR or multi-objective screening would broaden its applicability.
- Closed-loop discovery with lab feedback: Currently, the pipeline is one-directional – it produces ranked candidates. In practice, linking it to experimental feedback (e.g., active learning [25]) could create a self-improving cycle, where each new assay result refines the models and improves candidate prioritization.
Our work shows that early-stage drug discovery can be reimagined as an automated, modular workflow powered by machine learning. While the path from ranking molecules on a screen to delivering safe and effective medicines remains long, each advance in automation and integration brings us closer to faster, more affordable, and more reliable discovery.
[1] Tamimi, Nihad AM, and Peter Ellis. “Drug development: from concept to marketing!.” Nephron Clinical Practice 113.3 (2009): c125-c131.
[2] Wouters, Olivier J., Martin McKee, and Jeroen Luyten. “Estimated research and development investment needed to bring a new medicine to market, 2009-2018.” Jama 323.9 (2020): 844-853.
[3] Yamaguchi, Shingo, Masayuki Kaneko, and Mamoru Narukawa. “Approval success rates of drug candidates based on target, action, modality, application, and their combinations.” Clinical and Translational Science 14.3 (2021): 1113-1122.
[4] Hingorani, Aroon D., et al. “Improving the odds of drug development success through human genomics: modelling study.” Scientific reports 9.1 (2019): 18911.
[5] Dreiman, Gabriel HS, et al. “Changing the HTS paradigm: AI-driven iterative screening for hit finding.” SLAS DISCOVERY: Advancing the Science of Drug Discovery 26.2 (2021): 257-262.
[6] Claude code, https://www.anthropic.com/claude-code
[7] Paananen, Jussi, and Vittorio Fortino. “An omics perspective on drug target discovery platforms.” Briefings in bioinformatics 21.6 (2020): 1937-1953.
[8] Buniello, Annalisa, et al. “Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery.” Nucleic acids research 53.D1 (2025): D1467-D1475.
[9] Gaulton, Anna, et al. “ChEMBL: a large-scale bioactivity database for drug discovery.” Nucleic acids research 40.D1 (2012): D1100-D1107.
[10] Landrum, Greg. “Rdkit documentation.” Release 1.1-79 (2013): 4.
[11] Chen, Tianqi, and Carlos Guestrin. “Xgboost: A scalable tree boosting system.” Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining. 2016.
[12] Akiba, Takuya, et al. “Optuna: A next-generation hyperparameter optimization framework.” Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining. 2019.
[13] Irwin, John J., et al. “ZINC20—a free ultralarge-scale chemical database for ligand discovery.” Journal of chemical information and modeling 60.12 (2020): 6065-6073.
[14] Willett, Peter. “Similarity-based virtual screening using 2D fingerprints.” Drug discovery today 11.23-24 (2006): 1046-1053.
[15] Sheridan, Robert P., et al. “Similarity to molecules in the training set is a good discriminator for prediction accuracy in QSAR.” Journal of chemical information and computer sciences 44.6 (2004): 1912-1928.
[16] Lipinski, Christopher A. “Lead-and drug-like compounds: the rule-of-five revolution.” Drug discovery today: Technologies 1.4 (2004): 337-341.
[17] Zhou, Hongyi, and Jeffrey Skolnick. “Utility of the Morgan fingerprint in structure-based virtual ligand screening.” The Journal of Physical Chemistry B 128.22 (2024): 5363-5370.
[18] Bajusz, Dávid, Anita Rácz, and Károly Héberger. “Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?.” Journal of cheminformatics 7.1 (2015): 20.
[19] 90% of drugs fail clinical trials, https://www.asbmb.org/asbmb-today/opinions/031222/90-of-drugs-fail-clinical-trials
[20] Wang, Jiang, and Hong Liu. “Application of nitrile in drug design.” Chinese Journal of Organic Chemistry 32.9 (2012): 1643.
[21] Kalgutkar, Amit S., and Deepak Dalvie. “Predicting toxicities of reactive metabolite–positive drug candidates.” Annual review of pharmacology and toxicology 55.1 (2015): 35-54.
[22] Jiang, Dejun, et al. “Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models.” Journal of cheminformatics 13.1 (2021): 12.
[23] Van De Waterbeemd, Han, and Eric Gifford. “ADMET in silico modelling: towards prediction paradise?.” Nature reviews Drug discovery 2.3 (2003): 192-204.
[24] Ramsay, Rona R., et al. “A perspective on multi-target drug discovery and design for complex diseases.” Clinical and translational medicine 7.1 (2018): 3.
[25] Reker, Daniel, and Gisbert Schneider. “Active-learning strategies in computer-assisted drug discovery.” Drug discovery today 20.4 (2015): 458-465.
Learn more